Pues así, como si nada, ya se nos fue una tercera parte de este año. Este es mi tercer año ya ya fijo en esta ciudad. Y creo que he logrado muchas de las metas que me he trazado. Todavía faltan muchas, pero hay que tener paciencia. Poco a poco estoy creando presencia en el medio. Hace dos años nadie sabía que existía y aun así tuve trabajo. El número de pacientes y de casos va en aumento. Me sorprende como todavía después de tres años todavía hay quienes creen que vivo en otra ciudad, que solo vengo los fines de semana. Pero bueno. Debo ser paciente.
Lo importante es que en este tiempo ya logré identificar los principales problemas de salud de la región y ya sé en que debo enfocarme:
Hernias de disco
Hemorragia cerebral: llámese hipertensiva o subaracnoidoea
Tumores cerebrales
En ese orden. También ya identifiqué lo que la gente prefiere y como le gusta que los abordes al momento de ofrecerle la cirugía. Ya tropecé al momento de cobrar honorarios, es parte del proceso de aprendizaje, estoy aprendiendo eso de hacer “paquetes”, es natural cometer errores. Ya aprendí a trabajar con las compañías de seguros. Sé que en futuro vienen cosas buenas. Es cuestión de ser paciente y no volver a desesperar…
Un tumor cerebral es un crecimiento descontrolado de células derivadas de componentes cerebrales (tumores primarios) o de células tumorales localizadas en otras áreas del organismo (metástasis).1
Los tumores pueden ser benignos o malignos, dependiendo de la rapidez de su crecimiento y de si logran resecarse o curarse mediante el tratamiento neuroquirúrgico. A diferencia de los tumores de otros tejidos, la distinción entre manifestaciones benignas y malignas no es tan clara, por ejemplo, algunas lesiones benignas pueden infiltrar regiones enteras con comportamiento clínico maligno. Las neoplasias malignas producen metástasis, lo cual constituye un hecho excepcional. Las metástasis hacia el SNC provienen, en orden de frecuencia, del pulmón, mama, piel (melanoma), riñón y gastrointestinal y tienden a crecer entre la unión de la corteza y la sustancia blanca.
Clasificación
Determinación de la malignidad según las características histológicas del tumor.2
Tumores de evolución lenta (Bajo grado)
Tumores de grado I benignos, de crecimiento lento y circunscriptos.
Tumores de grado II De crecimiento lento, pero con límites imprecisos, o de extensión.
Tumores de evolución rápida (alto grado)
Tumores de grado III Tumores anaplásicos, su evolución es más rápida.
Tumores de grado IV Tumores malignos, muestran signos histológicos de crecimiento muy rápido en todas las regiones examinadas.
Tumores primarios
1. Gliomas
a. Astrocitomas
Constituyen el 25-30 por ciento de los gliomas. Se presentan en los hemisferios cerebrales en los adultos y en el cerebelo en los niños.
Glioblastoma multiforme, corte histológico.
Clasificación de la OMS para los astrocitomas
Grado I-OMS: Astrocitoma pilocítico, Tumor disembrioplástico neuroepitelialGrado II-OMS: Astrocitoma difuso (de bajo grado)
Grado III-OMS: Astrocitoma anaplásico
Grado IV-OMS: Glioblastoma multiforme
b. Oligodendroglioma
Este tipo de tumor representa el 6 por ciento de los gliomas, y suele presentarse entre la cuarta y quinta décadas de la vida. Su localización más frecuente es en los lóbulos frontal y temporal.4
Gliomas malignos
Son los tumores cerebrales primarios más frecuentes y agresivos. Causan globalmente un 2 por ciento de las muertes por cáncer.5 Los tres tipos histopatológicos más frecuentes son el glioblastoma multiforme, el astrocitoma anaplásico y el oligodendroglioma anaplásico. Estos tumores poseen una alta tasa de recidiva local tras tratamiento quirúrgico, progresando localmente, lo que finalmente termina causando la muerte del paciente. Se diseminan principalmente a través de la sustancia blanca o por vía líquido cefalorraquídeo. A diferencia del resto de las neoplasias malignas avanzadas, éstas no dan metástasis a distancia.
Glioblastoma multiforme: Constituye la forma más agresiva de los astrocitomas (tumor grado IV-OMS). Tradicionalmente se admitía que presentaba una supervivencia media, a los dos años, de aproximadamente 10 por ciento.6 El tratamiento requiere un abordaje multidisciplinar que incluye cirugía, radioterapia y quimioterapia. Con uno de estos protocolos de tratamiento, el protocolo de Stupp7 se ha alcanzado recientemente una supervivencia del 25% y aún más en los casos en los que se consiguió una extirpación completa. El ideal es que en el tratamiento participen especialistas en neurocirugía, neurología y oncología.8 Aunque el pronóstico es malo se han conseguido avances en los últimos años. Por ejemplo, la utilización de un producto (5-aminolevulinico) consigue colorear el tumor bajo luz fluorescente y mejorar la extirpación.9 Es recomendable impulsar la participación en ensayos clínicos para buscar nuevos tratamientos,10 se continúan buscando nuevos tratamientos con fármacos, inmunoterapia y virus oncolíticos.
2. Meningiomas
Constituyen el 15-20 por ciento de los tumores intracraneanos primarios. Son más frecuentes entre los 20 y los 60 años de edad. Se originan en las células aracnoideas de las meninges, son muy frecuentes y benignos; están encapsulados y bien limitados, aparecen en cualquier lugar del cerebro (supra e infratentorial).
Tumores secundarios (metástasis)
Las células cancerígenas de un sitio primario pueden viajar hasta el cerebro a través del sistema circulatorio, a través de la vía linfática y del líquido cefalorraquídeo. La forma más común es la circulatoria.
El cerebro es el sitio de preferencia de metástasis del melanoma y del cáncer de las células pequeñas de pulmón. En el varón, las metástasis provienen principalmente del pulmón, del colon y del riñón. En la mujer, los casos más frecuentes son el cáncer de mamas, de pulmón, de colon y el melanoma.11 Las metástasis espinales ocurren en el 5 por ciento de los pacientes con cáncer, más frecuentemente en el cáncer de mama, de próstata y elmieloma múltiple.
Clínica
Los tumores cerebrales causan síntomas variados. En general, se distinguen las manifestaciones derivadas de la hipertensión intracraneal, y los síntomas secundarios a la expansión tumoral, estos últimos denominadossignos focales, que dependen de la estructura anatómica afectada.
Síntomas de hipertensión intracraneal
Visión doble
Dolor en una extremidad
Cefalea
Vómitos
Edema de papila y alteraciones visuales
Trastornos del comportamiento (irritabilidad, labilidad emocional, fallos en el discernimiento, alteraciones de la memoria, falta de iniciativa, indiferencia a las costumbres sociales.)
Síndromes focales: Son manifestaciones que orientan la localización de la lesión.
Diagnóstico
El diagnóstico se realiza mediante exámenes imagenológicos como la TAC o la resonancia magnética (RM), las cuales permiten conocer la localización y el tamaño del tumor y además sugerir la naturaleza del mismo, pero es la biopsia la que indica el tipo exacto de tumor.
El 99% de los casos de acromegalia son por causa del aumento de hormona de crecimiento (GH) originada por tumores de la hipófisis (generalmente macroadenomas productores de GH) monoclonales con activación de oncogenes GSP (proteína G estimuladora). En el 1%, se presenta por paraneoplasia con liberación de FSH-RH. Su frecuencia va descendiendo. Evolución lenta.
Diagnóstico
El diagnóstico continúa siendo tardío a pesar de que físicamente los pacientes inician los signos y síntomas hasta ocho años previos al diagnóstico.
Se hace muchas al observar la evolución estudiando fotos antiguas (10-20 años). Si el inicio de la liberación excesiva de hormona de crecimiento es postpuberal produce acromegalia; si es prepuberal, gigantismo.
Características físicas de la acromegalia: cara: separación de los dientes, prognatismo, macroglosia, rasgos toscos, aumento del arco supraciliar, aumento del tamaño de la nariz, labios, aumento de los surcos de la cara. A otros niveles: artralgias, aumento de partes blandas, manos y pies, debilidad muscular, hipertensión arterial (HTA), síndrome del túnel carpiano, voz gruesa, organomegalia (por ejemplo: glándula tiroides), diaforesis (sudoración) excesiva, trastorno de los ciclos menstruales, impotencia (50%).
En pacientes con acromegalia, la prevalencia de determinadas neoplasias benignas y malignas es superior que en la población sana. El macroadenoma de hipófisis y microadenoma se aprecia en el 63,4% y 25,7% de estos pacientes, respectivamente.
Los niveles promedio de GH e IGF-1 antes de la neurocirugía fueron 20,2 (IQR = 34,9) ng / ml y 764,5 (IQR = 569,6) ng / ml, respectivamente.
Se han encontrado las siguientes prevalencias de diversos tumores: bocio nodular – 64/101 pacientes (63,0%), pólipos del colon – 13/101 pacientes (13,0%), pólipos uterinos – 4 / 101 pacientes (4,0%) y adenoma de próstata – 2 / 101 pacientes (2,0%). Entre los tumores malignos, el cáncer de tiroides, de endometrio y cáncer de cuello uterino fueron las más frecuentes, cada uno de estos se producen en 3 pacientes (3,0%).
La prevalencia del cáncer de colon fue de 2,0% (en 2 pacientes).
Se sugiere un aumento global de la incidencia de tumores en pacientes con acromegalia, por lo que son necesarios futuros estudios multicéntricos para resolver el significado de esta observación (Bałdys 2010).
En pacientes con acromegalia, la intensidad de la señal T2 en Resonancia en el momento del diagnóstico se correlaciona con las características histológicas y bioquímicas y predice el resultado de la primera línea de tratamiento (Heck y col., 2011).
Pronóstico
Esta enfermedad tiene una elevada tasa de mortalidad (1.5 a 3 veces en comparación a la tasa de la población general) casi siempre por causa cardio o cerebrovascular, reduciendo la esperanza de vida de la persona que la padece en por lo menos 10 años.
Bibliografía
Bałdys-Waligórska, Agata, Anna Krzentowska, Filip Gołkowski, Grzegorz Sokołowski, y Alicja Hubalewska-Dydejczyk. 2010. The prevalence of benign and malignant neoplasms in acromegalic patients. Endokrynologia Polska 61, no. 1 (Febrero): 29-34.
Heck, Ansgar, Geir Ringstad, Stine Lyngvi Fougner, Olivera Casar-Borota, Terje Nome, Jon Ramm-Pettersen, and Jens Bollerslev. 2011. “Intensity of pituitary adenoma on T2 weighted MRI predicts the response to octreotide treatment in newly diagnosed acromegaly.” Clinical Endocrinology (November 8). doi:10.1111/j.1365-2265.2011.04286.x.http://www.ncbi.nlm.nih.gov/pubmed/22066905.
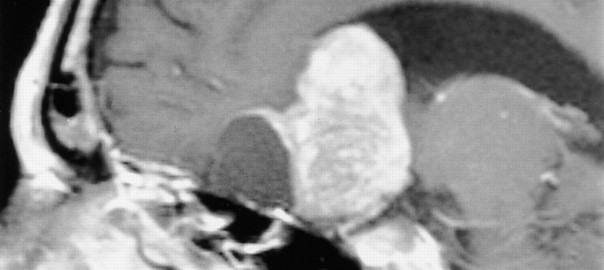
Tumores benignos pero localmente invasivos de la parte anterior (rara vez posterior) de la glándula hipofisaria alojada en la silla turca.
Epidemiología
La incidencia, en series de autopsia alcanza hasta un 25% de la población. En orden decreciente, los adenomas hipofisarios son el tercer tipo de tumor primario del sistema nervioso central, sumando casi el 17 % de las neoplasias intracraneales, afectando por igual a hombres y mujeres en la tercera y cuarta década de la vida (Couldwell 2009).
Los adenomas hipofisarios son extremadamente raros en niños prepúberes (Furtado 2010).
Los no funcionantes son mas frecuentes que los funcionantes y predominan los prolactinomas dentro de los funcionantes.
Coexistencia de aneurismas
La edad avanzada y la existencia de una invasión del seno cavernoso se correlacionaron con una mayor incidencia de aneurismas. En la serie de Oh y col., hasta en un 2.3% (Oh y col., 2012).
Sasagawa y col., 6,9 % (Sasagawa y col., 2012).
Clasificación
En cuanto a su tamaño clasificamos los tumores como microadenomas (menos de 10 mm.) y macrodenomas.
Los adenomas hipofisarios gigantes son mayores de 4 cm.
Clasificación hormonal
Según la secreción hormonal en funcionante o no funcionante y el tipo de hormona que producen:
Adenoma productor de GH (Acromegalia)
Adenoma productor de FSH
Adenoma productor de LH
Adenoma productor de PRL
Adenoma productor de TSH
Adenoma productor de ACTH
Etiología
La causa de la mayoría de los tumores hipofisarios sigue siendo desconocida, aunque la contribución genética es reconocida por algunos (Couldwell 2009).
La expresión de la survivina, tanto en células normales, como en tumores de hipófisis sugiere que puede jugar un papel importante en la regulación de la proliferación de la glándula (Wasko 2009).
Pueden estar asociados al síndrome de neoplasia endocrina múltiple ( NEM ) , especialmente el tipo I. ( a diferenciar con el síndrome de Zollinger-Ellison,) hiperparatiroidismo.
Localización
Su origen es principalmente a partir de la adenohipófisis ; los tumores de la neurohipófisis son raros.
Clínica
Generalmente se expresa por síntomas visuales y cefaleas referida a la región temporal, frontal occipital o retroocular.
Síntomas hormonales:( amenorrea- galactorrea, pérdida de la libido, erección, acromegalia o síndrome de Cushing…).
Con menor frecuencia trastornos de pares craneales ( III, IV, VI )
En la acromegalia:
Crecimiento de manos, de pies, y de huesos faciales; dolor, apnea del sueño; sudor excesivo.. Estos cambios son graduales y a menudo diferenciados por familiares del paciente y atribuido a envejecimiento.
La apoplejía hipofisaria es un síndrome clínico agudo que se puede presenta en los adenomas de hipófisis caracterizado por cefalea de inicio rápido, vómitos, disminución de la agudeza y/o campo visual, oftalmoplejía y disminución del nivel de conciencia. Este síndrome está causado por el infarto isquémico o hemorrágico de la glándula hipofisaria.
La RM es la prueba diagnóstica de elección.
Diagnóstico
Desde que se dispone de la RM cerebral se diagnostica en el 10 % de los pacientes de forma incidental (incidentaloma).
Pero la localización de microadenomas en la enfermedad de Cushing puede ser difícil, ya que hasta en un 45% de los pacientes la RM selar no es capaz de detectarla aunque la ecografía intraoperatoria transesfenoidal puede identificarlo como estructuras hiperecoicas (Knappe 2009).
EXPLORACION
Una vez exista la sospecha diagnóstica, se debe realizar un examen clínico con énfasis en :
SÍNTOMAS VISUALES : Agudeza visual Evaluación de campos visuales Compromiso de pares craneales ( III, IV, VI ) (Motilidad ocular).
Disfunción hipofisaria.
Pruebas diagnósticas necesarias:
RNM sin y con Gadolinio : La resonancia magnética simple y contrastada es el método diagnóstico de elección en todo paciente con sospecha de lesión de hipófisis. Establece los límites y la extensión, muestra las estructuras comprometidas y en algunos casos puede ayudar a determinar la consistencia del tumor. Así mismo es capaz de diagnosticar la apoplejía hipofisaria.
Exámenes de laboratorio
Endocrinos : PRL < 25 Normal 25-150 Hipotiroidismo primario Efecto de presión sobre el tallo Prolactinoma ? ? ?
150 Prolactinoma
ACTH Cortisol 8 a.m. 6-18 ug / 100 ml Supresión de dexametosa a dosis bajas : <5 ug /dl no Cushing 5 – 10 indeterminado, repetir 10 probable Cushing
Pruebas especializadas ACTH :
Supresión a dosis bajas por dos días Estimulación con cosyntropin Tolerancia a la insulina
Pruebas para diferenciar enfermedad de Cushing de producción ectópica y tumores adrenales :
Supresión dexametasona a dosis altas Estimulación con CRH Muestreo hormonal en sangre de senos petrosos
T3-T4-TSH Aumentan todas en adenomas productores ( muy raros ) Aumento de TSH y disminución de T4 : Hipotiroidismo primario Crecimiento de hipófisis secundario Disminución de TSH y T4: Hipotiroidismo secundario Compresión tumoral
FSH – LH, TESTOSTERONA Y ESTRADIOL En mujeres diagnostico difícil porque aumentan en peri y postmenopausia.
HORMONA DEL CRECIMIENTO VN en ayuno < 5 ng / ml Acromegalia 10 ng / ml Secreción por picos Pruebas especializadas : Somatomedina C Supresión con glucosa Estimulación con GNRH
Diagnóstico diferencial
Aneurisma intraselar
La presencia de un aneurisma intracraneal junto con un adenoma presenta gran riesgo de hemorragia subaracnoidea, durante la cirugía, particularmente cuando el aneurisma se encuentra cerca del campo operatorio, por lo que el aneurisma cerebral se trata primero (Yamada y col., 2012).
Craneofaringioma
Cordoma
Metástasis
Quiste de la bolsa de Rathke
Quiste aracnoideo
Gangliocitoma
En una serie de tratamiento quirúrgico por vía transesfenoidal de 300 casos, 29 lesiones selares no fueron adenomatosos (9,7%. Retrospectivamente la presentación clínica fue generalmente la de efecto de masa local. La extirpación total del tumor por vía transesfenoidal se logró en 17 casos y no fué necesario ningún tratamiento suplementario.
Las lesiones selares no adenomatosos representan una entidad clínica más frecuente de lo esperado, por lo que se debe tener presente su existencia (Koutourousiou 2010).
Tratamiento
Debe ser flexible y acomodado a cada paciente en particular.
Ningún especialista médico tiene suficientes conocimientos para proporcionar el cuidado completo de estos pacientes.
No es apropiado realizar cirugía en pacientes sin obtener una evaluación preoperatoria por el endocrinólogo.
No es aceptable intervenir quirúrgicamente y después referirlos para evaluación endocrina.
Se realiza de forma multidisciplinaria: Médico del paciente, Endocrinología, Oftalmología, Neurocirugía, Neurorradiología y Radioterapia.
Tratamiento médico
Tumores productores de PRL
En individuos con PRL 150 el tumor es usualmente un prolactinoma o un tumor plurihormonal productor de PRL, los cuales tienen muy buena respuesta a los agonistas dopaminérgicos. Se usa Bromocriptina iniciando a dosis bajas 2.5 mg / día para así disminuir la aparición de efectos secundarios, incrementándose según la respuesta hasta un máximo de 7.5 mg / día. El manejo inicial de los prolactinomas gigantes es médico y solo en caso de no haber respuesta se realizaría un tratamiento quirúrgico (Furtado 2010).
Tumores productores de hormona del crecimiento
Se utiliza Octeotrido el cual es un análogo de la somatostatina , es efectivo en el 90% de los pacientes con acromegalia, desafortunadamente su costo es muy alto, por lo que su utilidad práctica sería darlo desde los 15 días antes de la cirugía con el objetivo de disminuir el tamaño tumoral y así hacer mas fácil la resección.
La cabergolina no ha resultado eficaz y sólo se puede considerar en tumores pequeños y representación clínica leve (3).
Tratamiento quirúrgico
Con excepción del prolactinoma, el tratamiento de primera línea para la mayoría de los adenomas,es la resección quirúrgica; la cual puede ser a través del cráneo o seno esfenoidal (Sandoval-Sánchez y col., 2007).
Indicación quirúrgica:
Apoplejía hipofisiaria Deterioro visual rápido y/o progresivo Tumores productores de ACTH y TSH Productores de gonadotrofinas Productores de H. del crecimiento ( Manejo médico difícil por el costo económico. ) Areas hemorrágicas y necróticas en imágenes. Deseo del paciente Prolactinomas que no se controlan con bromocriptina,cabergolina o quinagolida o que producen efectos secundarios indeseables. Todos los tumores que producen efecto masa. En aquellos casos en que el diagnóstico es incierto.
Seno esfenoidal
En el caso de las rutas hacia el seno esfenoidal, se han descrito las vías transantral, transmaxilar, sublabial y endonasal.
Las dos primeras rara vez se emplean en la actualidad, mientras que la endonasal transesfenoidal es la ruta preferida para más del 90% de las lesiones de la silla turca.
En el abordaje endonasal transeptal transesfenoidal, se realiza una incisión en la columnela a nivel de la unión de la piel con la mucosa nasal, y con ello una amplia disección de dicha mucosa en el plano subpericondrial.
Endonasal directo transesfenoidal (Griffith y Veerapen, 1987).
Requiere mínima disección de la mucosa nasal, ya que se efectúa una incisión vertical más posterior, esto es, a nivel de la unión del cartílago septal con el septum óseo y a partir de ahí se realiza la disección, para luego luxar el cartílago septal e identificar el vómer, sin embargo, este abordaje provee un trayecto ligeramente fuera de la línea media.
A pesar de la utilización, cada vez mayor, del abordaje endonasal directo transesfenoidal, en la resección de adenomas de hipófisis, existen pocos estudios que describan la frecuencia de complicaciones o molestias postoperatorias, relacionadas con este abordaje.
Ha sido, desde la segunda mitad del pasado siglo, la alternativa de solución más versátil y utilizada, alcanzando más del 95% de las operaciones cuyo blanco es la silla turca.
El uso del endoscopio en la cirugía pituitaria a través de los senos paranasales fue reportado por vez primera en los años 1970 en la Literatura médica alemana. En 1989, Papay emplea el abordaje transeptal endoscópico para reparar fístulas de l.c.r. secundarias a cirugía pituitaria y en 1992, Jankowski reporta la resección endonasal endoscópica exitosa de adenomas hipofisarios en tres pacientes. Dos años después, Gamea expone su experiencia en 10 casos de tumores hipofisarios abordados por vía sublabial transeptal transesfenoidal utilizando el microscopio quirúrgico y apoyándose con el endoscopio. Este autor concluyó que el endoscopio facilitaba la disección, del tumor de la glándula normal.
En 1996, Sethi publica una serie de 40 pacientes tratados consecutivamente mediante la aplicación del abordaje transnasal endoscópico para el tratamiento de adenomas hipofisarios y craneofaringiomas y en el mismo año y el siguiente Jho y Carrau publicaron en sendos artículos, su experiencia inicial primero y luego en 50 pacientes, con ayuda de otro cirujano que sostiene el endoscopio después de la esfenoidotomía y le permite la operación bimanual. Inmediatamente reportan la introducción de un soporte mecánico con este fin.
Según Rodziewicz y Heilman, la descompresión endoscópica de los tumores pituitarios puede realizarse alcanzando buenos resultados con mínima morbilidad quirúrgica.
Aldo Stamm señala varias ventajas de la cirugía pituitaria endonasal endoscópica. La primera y más importante es proveer un acceso más directo y rápido a la silla sin craneotomía, lo que reduce la morbilidad asociada a este tipo de proceder. También mejora la visualización, el ángulo visual, y amplía la perspectiva panorámica de importantes estructuras anatómicas del seno esfenoidal, silla turca y región paraselar, permitiendo un manejo dinámico y con seguridad durante la cirugía.
Algunos autores han llegado a plantear que el endoscopio ha reemplazado al microscopio quirúrgico en la cirugía de los adenomas hipofisarios.
El aporte del grupo de la Universidad Federico II de Napóles, trabajando sobre la anatomía endoscópica y el diseño de instrumentos adecuados para esta cirugía, ha sido sustancial con vistas a convertirla en una técnica estándar (4, 10).
En determinados pacientes la radiocirugía estereotáctica puede ser eficaz (12), especialmente en aquellos pacientes no subsidiarios de tratamiento médico o quirúrgico (5), o en casos de restos tumorales a nivel de seno cavernoso (7).
Pauta medicamentosa preoperatoria:
Hidrocortisona 50 mg I.M a las 23 horas y a las 6 de la mañana. Antes de bajar al quirófano: Suero de 1000 ml + 20 mEq. + 50 mg. hidrocortisona 75 ml/hora.
Pauta intraoperatoria y postoperatoria:
Antibióticos ( Cefalotina 1 gm IV c/6h por 2 días, luego pasar a cefalexina VO 1 gm/6h por 5 días.) 100 mg. Hidrocortisona e.v cada 8 horas 2 día 100 mg IV c/12h 3 día 100 mg IV día 4 día Suspender
Control de ingesta, eliminación. Electrolitos diarios hasta el segundo día, BUN, glicemia, osmolaridad. Manejo de las posibles complicaciones (Diabetes insípida )
Postoperatorio
Se envía la muestra patológica para inmunohistoquímica y microscopía electrónica . Se toma cortisol plasmático al sexto día, si es < de 5 repetir en una semana, si continua < 5 hacer prueba de estímulo con ACTH ; si persiste bajo se debe iniciar reemplazo hormonal. A los quince días se toma PRL si es alta se debe iniciar Bromocriptina. RM de control a los tres meses. Control de tamizaje hormonal a los seis meses.
Radioterapia/Radiocirugía
La radioterapia es un tratamiento establecido para tumores recurrentes y recidivas.
La radiocirugía recientemente ha mostrado un control tumoral para tumores residuales y recidivas de adenomas de hipófisis.
Recidiva
En caso de recidiva el manejo ideal es la reintervención Utilizamos radioterapia o radiocirugía estereotáxica en caso de tumores productores de hormona del crecimiento que a los 15 días postoperatorios el control hormonal persista alto y el paciente no pueda tomar Octeótrido y no exista posibilidad de una segunda intervención Pacientes con tumores productores y no productores de PRL en los que exista recidiva y no sea posible la resección quirúrgica ( tumor inoperable, no respuesta a cirugía ni a bromocriptina, no aceptación del paciente ).
Algunos tumores recidivantes se han tratado con quimioterapia BCNU e incluso recientemente con Gliadel.
Gliadel for Pituitary Adenomas and Craniopharyngiomas Edward R. Laws, Jr., M.D., F.A.C.S.; Angel M. Morris, R.N., B.S.N., C.C.R.N., C.C.R.C.; Nicholas Maartens, M.D.Department of Neurosurgery, University of Virginia Health Sciences Center, Charlottesville, Virginia Department of Neurosurgery, University of Virginia Health Sciences Center, Charlottesville, Virginia Department of Neurosurgery, The Royal Melbourne Hospital, Melbourne, Australia.
Las fístulas de líquido cefalorraquídeo (l.c.r.) a través del piso selar consecutivas a la cirugía o espontáneas (casi siempre en relación con silla vacía), tampoco son infrecuentes en la práctica neuroquirúrgica.
La terapia génica en el campo experimental es desalentador quizá debido a la pobre difusión de virus vectores en el tejido hipofisario (2).
Pronóstico.-
Tasa de recidiva del 20 % a pesar de una resección aparentemente completa y que depende de su característica histológica especialmente los acidófilos, los adrenocorticotrópicos, oncocitomas, plurihormonales y tumores con indices proliferativos inusualmente altos.
Prolactinomas.- La mortalidad quirúrgica en series recientes es<0.5% y curaciones de prolactinomas a largo plazo > del 74%. Resultados similares al tratamiento médico pero evitando los efectos secundarios del tratamiento médico.
Acromegalia
El embarazo en pacientes con acromegalia es un evento poco frecuente, debido a la función perturbada de gonadotropos. Por otra parte, el embarazo puede provocar un aumento del tamaño del adenoma o un aumento de la secreción de la hormona del crecimiento (GH). en En mujeres jóvenes bien controlados con tratamiento médico el embarazo no suele influir en la acromegalia (Ben Salem 2009).
Bibliografía
Ben Salem Hachmi L, Kammoun I, Bouzid C, Smida H, Nagi S, Turki Z, Ben Slama C. [Management of acromegaly in pregnant woman.]. Ann Endocrinol (Paris). 2009.
Carri NG, Sosa YE, Brown OA, Albarino C, Romanowski V, Goya RG: Studies on in vivo gene transfer in pituitary tumors using herpes-derived and adenoviral vectors. Brain Res Bull 65:17-22, 2005.
Couldwell WT, Cannon-Albright L. A heritable predisposition to pituitary tumors. Pituitary 2009.
Freda PU, Reyes CM, Nuruzzaman AT, Sundeen RE, Khandji AG, Post KD: Cabergoline therapy of growth hormone & growth hormone/prolactin secreting pituitary tumors. Pituitary 7:21-30, 2004.
Furtado SV, Saikiran NA, Ghosal N, Hegde AS. Giant, solid, invasive prolactinoma in a prepubescent boy with gynecomastia. Pediatr Neurol. 2010 Jan;42(1):72-4.
Gonzalez-Gonzalez JL, Lopez-Arbolay O, Morales-Sabina O, Marti-Pineiro JF, Vidal-Verdial R: [Transnasal-transsphenoidal endoscopic surgery of the sellar region]. Neurocirugia (Astur) 16:27-33, 2005.
Jane JA, Jr., Vance ML, Woodburn CJ, Laws ER, Jr.: Stereotactic radiosurgery for hypersecreting pituitary tumors: part of a multimodality approach. Neurosurg Focus 14:e12, 2003.
Knappe UJ, Engelbach M, Konz K, Lakomek HJ, Saeger W, Schönmayr R, Mann WA.Ultrasound-assisted Microsurgery for Cushing’s Disease. Exp Clin Endocrinol Diabetes.
Koutourousiou M, Kontogeorgos G, Seretis A. Non-adenomatous sellar lesions: experience of a single centre and review of the literature. Neurosurg Rev. 2010 May 18. [Epub ahead of print] PubMed PMID: 20480381.
Kreutzer J, Fahlbusch R: Diagnosis and treatment of pituitary tumors. Curr Opin Neurol 17:693-703, 2004.
Liu JK, Schmidt MH, MacDonald JD, Jensen RL, Couldwell WT: Hypophysial transposition (hypophysopexy) for radiosurgical treatment of pituitary tumors involving the cavernous sinus. Technical note. Neurosurg Focus 14:e11, 2003.
McCord MW, Buatti JM, Fennell EM, Mendenhall WM, Marcus RB, Jr., Rhoton AL, Grant MB, Friedman WA: Radiotherapy for pituitary adenoma: long-term outcome and sequelae. Int J Radiat Oncol Biol Phys 39:437-444, 1997.
Oh, Min Chul, Eui Hyun Kim, and Sun Ho Kim. 2012. “Coexistence of Intracranial Aneurysm in 800 Patients with Surgically Confirmed Pituitary Adenoma.” Journal of Neurosurgery 116 (5) (May): 942–947. doi:10.3171/2011.12.JNS11875.
Pollock BE, Carpenter PC: Stereotactic radiosurgery as an alternative to fractionated radiotherapy for patients with recurrent or residual nonfunctioning pituitary adenomas. Neurosurgery 53:1086-1091; discussion 1091-1084, 2003.
Rudnik A, Zawadzki T, Wojtacha M, Bazowski P, Zubgaluszka-Ignasiak B, Duda I: [Endoscopic transsphenoidal treatment of pituitary adenomas.]. Neurol Neurochir Pol 39:17-23, 2005.
Sandoval-Sánchez, J H, F Flores-Cárdenas, M C Vargas-Frutos, y J M Páez-Ontiveros. 2007. «[Complications of the direct endonasal transsphenoidal approach in the management of pituitary adenomas]». Neurocirugía (Asturias, Spain) 18 (6) (Diciembre): 485-491.
Sasagawa, Yasuo, Osamu Tachibana, Shunsuke Shiraga, Hisasi Takata, Takuya Akai, and Hideaki Iizuka. 2012. “[A Clinical Feature and Therapeutic Strategy in Pituitary Adenomas Associated with Intracranial Aneurysms].” No Shinkei Geka. Neurological Surgery 40 (1) (January): 15–21.
Sheehan JM, Vance ML, Sheehan JP, Ellegala DB, Laws ER, Jr.: Radiosurgery for Cushing’s disease after failed transsphenoidal surgery. J Neurosurg 93:738-742, 2000.
Sinha, Sumit, y B S Sharma. 2010. Giant pituitary adenomas–an enigma revisited. Microsurgical treatment strategies and outcome in a series of 250 patients. British Journal of Neurosurgery 24, no. 1 (Febrero): 31-39. doi:10.3109/02688690903370305.
Wasko R, Waligorska-Stachura J, Jankowska A, Warchol JB, Liebert W, Sowinski J. Coexpression of survivin and PCNA in pituitary tumors and normal pituitary. Neuro Endocrinol Lett. ;30(4)2009.
Yamada, So, Shoko M Yamada, Toshio Hirohata, Yudo Ishii, Katsumi Hoya, Mineko Murakami, and Akira Matsuno. 2012. “Endoscopic Extracapsular Removal of Pituitary Adenoma: The Importance of Pretreatment of an Adjacent Unruptured Internal Carotid Artery Aneurysm.” Case Reports in Neurological Medicine 2012: 891847. doi:10.1155/2012/891847.


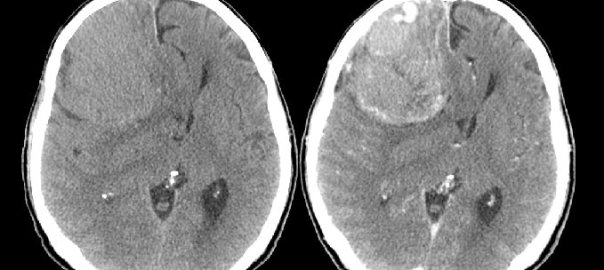


 El 99% de los casos de acromegalia son por causa del aumento de hormona de crecimiento (GH) originada por tumores de la hipófisis (generalmente macroadenomas productores de GH) monoclonales con activación de oncogenes GSP (proteína G estimuladora). En el 1%, se presenta por paraneoplasia con liberación de FSH-RH. Su frecuencia va descendiendo. Evolución lenta.
El 99% de los casos de acromegalia son por causa del aumento de hormona de crecimiento (GH) originada por tumores de la hipófisis (generalmente macroadenomas productores de GH) monoclonales con activación de oncogenes GSP (proteína G estimuladora). En el 1%, se presenta por paraneoplasia con liberación de FSH-RH. Su frecuencia va descendiendo. Evolución lenta.